
Recently, my coworkers and I put out a preprint “Classical solution of the FeMo-cofactor model to chemical accuracy and its implications’’ (Zhai et al. 2026). It is a bit unusual to write commentary on one’s own scientific article. However, in this case, given the many inquiries I have had about the work in the context of quantum computing, many of which have contained similar questions (and often similar misunderstandings), I thought it would be useful to provide some perspective that we could not provide in the original preprint, in an informal manner.
What is FeMo-co?
I will start with some background on the FeMo-cofactor (FeMo-co). This cofactor is the reaction center of nitrogenase, an enzyme found in certain soil-dwelling bacteria. Nitrogenase’s claim to fame is that it converts atmospheric dinitrogen, which is held together by a strong N-N triple bond, into a reduced form (ammonia) which can then be taken up by plants and thereby be passed onto the rest of the living biomass. In terms of incorporating nitrogen into biomass, nitrogenase is believed to be responsible for about 2/3 of biological nitrogen, with the remainder coming from fertilizers. Because it plays this critical role, it is sometimes referred to as the enzyme that feeds the planet.
The chemistry of how dinitrogen is reduced at the FeMo-cofactor is still largely unknown. The basic stoichiometry of the reaction is often written as

but this just a sketch of the process. In particular, the above equation contains, nominally, a large number of molecular reactants, and clearly they do not all just come together in a bang! The role of the cofactor, and the enzyme more generally, is to coordinate the protons, electrons, biological energy source (ATP), and the dinitrogen molecule, into a sequence of well-defined steps, known as the reaction mechanism. Since the work of Lowe and Thorneley (Thorneley and Lowe 1984), the most common proposal for the nitrogenase reaction mechanism contains 8 intermediate steps (corresponding roughly to 8 sequential proton and electron additions). However, due to the difficulty in isolating the intermediate states of FeMo-co, as well as challenges in using experimental probes to deduce what these states are, the Lowe-Thorneley cycle still remains an unproven hypothesis. Biochemists, spectroscopists, as well as a few theoretical quantum chemists, are today actively engaged in observing, computing, deducing (and arguing about) the nitrogenase mechanism (Jiang and Ryde 2023; Lancaster et al. 2011; Einsle and Rees 2020; Badding et al. 2023; Thorhallsson et al. 2019).
So how did nitrogenase become so widely discussed in the setting of quantum computing? In 2016, an article “Elucidating reaction mechanisms on quantum computers’’, that has since become one of the most cited papers in the nitrogenase field, arguably started this all (Reiher et al. 2017). The article included a number of proposals, including (1) that the ‘promise of exponential speedups for the electronic structure problem’ could be applied to elucidate the nitrogenase reaction mechanism that had so far proved intractable for classical computation, and (2) that solving this problem would be an example of how quantum simulation could be ‘scientifically and economically impactful’. (Similar proposals can also be found repeated in less technical language and settings, see e.g. ‘Why do we want a quantum computer’). An important technical contribution of the article was to provide a detailed quantum resource estimate for a simulation of chemistry. The problem statement was to compute the ground-state energy of a specific ‘54 orbital’ (108 qubit) model of FeMo-co, to an accuracy of 1 kcal/mol, referred to as chemical accuracy. It is important to note the word ‘model’ in the problem statement. Electrons move in continuous space, and thus quantum chemical Hamiltonians are formulated in the continuum, while quantum computation requires discretization of this space. This discretization, in terms of a so-called active space set of orbitals that the electrons can variously occupy, constitutes the model. We will return to the definition of the model below. By compiling a Trotter-Suzuki implementation of the quantum phase estimation algorithm within a fault-tolerant resource model for their specific FeMo-co model Hamiltonian, Ref. (Reiher et al. 2017) provided a T-gate resource estimate. Combined with some assumptions about the quantum architecture, this provided perhaps the first concrete time-cost to solve an interesting chemistry problem on a quantum computer. This work has since served as an inspiration for many subsequent quantitative resource estimation efforts in the quantum computing for chemistry field.
Exponential speedup and societal impact
Before proceeding further in this story, it is worth examining the two key propositions made in Ref. (Reiher et al. 2017). I start with the question of exponential speedup. Quantum algorithms for the ground-state energy, such as quantum phase estimation, essentially perform a projective measurement of the energy (encoded in a phase). Thus, it is essential to prepare a good initial state, i.e. with large overlap with the desired eigenstate, to measure the correct energy. This, however, is a strong constraint, if we are seeking asymptotically large quantum advantage. For example, if such an initial state is first determined classically, as is often suggested, then exponential quantum advantage in a given problem requires that finding good classical guesses is easy, while improving them classically to fully solve the problem becomes exponentially hard as the problem size increases. Unfortunately, convincing evidence that chemically relevant electronic structure problems, including the problem of cofactor electronic structure exemplified by FeMo-co, fall into this category has not yet been found, as discussed in detail in Refs. (Lee et al. 2023; Chan 2024).
The second proposition, that elucidating the reaction mechanism of nitrogenase will lead to a transformative societal impact, is similarly nuanced. The claim originates in the observation that the competing industrial process for fertilizer production via nitrogen reduction, namely, the Haber-Bosch process, takes place at high temperatures and pressures and consumes a significant percentage of the world’s energy. Bacteria, on the other hand, can do this process at room temperature.
While it is true that the nitrogenase enzyme functions at ambient temperature and pressure, it is simply false that it consumes much less energy. This is because the large amount of energy required for nitrogen fixation mainly originates from thermodynamics, i.e. one needs energy to break the strong nitrogen triple bond. In fact, taking into account the physiological conditions and the ATP cost, bacteria arguably expend more energy to reduce ammonia (Chan 2024) than a modern efficient industrial implementation of the Haber-Bosch process. Thus the real hope behind trying to understand the nitrogenase mechanism in the context of societal impact is that we may one day engineer a variant of it with more desirable properties, e.g. with higher turnover, or with a lower carbon footprint, or which is more selective for nitrogen reduction. Whether this is actually possible remains to be seen, and certainly requires much more than knowing the ground-state of FeMo-co, or even the full reaction mechanism.
Which FeMo-cofactor model?
I now return to the question of FeMo-cofactor models. Ref. (Reiher et al. 2017) introduced a particular cofactor model, which I will refer to it as RWST, following the names of the authors. As we soon found out, simulating the ground-state of the RWST model was actually very easy classically, and in fact (as reported in (Li et al. 2019)) could be done using standard quantum chemistry methods with a few hours of calculation on a laptop. This was because although the RWST model was a 108 qubit model, and (in the worst case) a 108 qubit ground-state cannot be stored classically, the RSWT model Hamiltonian was constructed in such a way to not capture any of the difficult features of the FeMo-cofactor ground-state. This highlights the importance of not assuming worst case complexity about physical problems!
What makes the electronic structure of the FeMo-cofactor (relatively) complicated is the presence of many ‘unpaired’ electrons. In simple molecules, we can describe the ground-state as one where all the electrons sit in pairs in orbitals. Since an orbital can only carry a pair of electrons at a time, the ground-state is simply described by filling the lowest energy orbitals with pairs. However, in molecules with transition metals, there are typically ‘unpaired’ electrons (so-called open-shells), and then we need to consider whether and how they pair up, which orbitals are singly versus doubly occupied, and so on. The RSWT model ground-state had no unpaired electrons! It was therefore unrepresentatively easy to solve for the ground state classically.
Because of the problems with the RWST model, my group formulated a more suitable 76 orbital/152 qubit model of FeMo-co in Ref. (Li et al. 2019), which I will refer to as the LLDUC model, again by the names of the authors. Although the LLDUC model is still a significant truncation of the true electronic structure of FeMo-co, we verified that it contains the correct open-shell character of the cofactor, and thus has a ‘representative’ complexity in its ground-state. Since we published the LLDUC model, it has become the most common benchmark model of FeMo-co used in quantum resource estimates for new quantum chemistry ground-state algorithms (Wan et al. 2022; Berry et al. 2019; Luo and Cirac 2025; Low et al. 2025).
Heuristics in the classical solution of the LLDUC FeMo-cofactor model
This brings me now to the recent work in Ref. (Zhai et al. 2026), where, through a sequence of classical calculations, we could produce a classical estimate of the ground-state energy of the LLDUC model to chemical accuracy. How was this achieved?
Classical electronic structure methods (aside from exact
diagonalization) are heuristic algorithms. Much like quantum algorithms,
they implicitly or explicitly start from an initial state. In chemical
applications, this can be viewed as a product state or set of product
states: for tensor network algorithms, such as the density matrix
renormalization group (when not considering topological order) this is
the set of 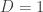

In FeMo-co, unlike in simpler molecules, it is not at all obvious
what product state to start from. To address this, in Ref. (Zhai et al.
2026), we devised an enumeration and filtering protocol. The
relevant manifold arises from the orbital and spin degrees of freedom of
the Fe ions: which Fe orbitals are occupied, by how many electrons, and
with which spins. One technical point is that the resulting product
states do not generally conserve the global 

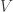

Because applying the highest level of classical approximation to all enumerated product states was far too expensive, we used a filtering funnel, where product states were ranked at different levels of theory, passing promising candidates to higher levels of classical computation. In the end, the final most accurate calculations were performed on only 3 candidates, which we deduced to all be essentially degenerate to within chemical accuracy.
There are other important technical details in Ref. (Zhai et al. 2026) which I have not mentioned: the use of unrestricted orbitals, the systematic extrapolations to obtain the final energies and estimated errors, and the benchmarking required to be confident about the protocol. However, recognizing that the FeMo-co ground-state problem could be reduced essentially to a ranking problem was the essence of what made the estimate possible.
Implications of the classical solution for chemistry
From a chemical and biochemical perspective, computing the
ground-state energy of a model to some specified accuracy – even
chemical accuracy – is a highly artificial target. Most chemical
calculations that have an impact on our understanding never achieve or
even target chemical accuracy in the total energy. In addition,
chemistry does not depend on the the total energy, but the relative
energy of different chemical configurations, which typically differ only
by 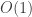
The main take-home from our work then is that there is nothing especially mysterious about FeMo-co’s electronic structure. The story of the FeMo-co ground-state is not one of multiconfigurational electronic structure (i.e. where the states are not at all close to product states), but one of multiple configurations (i.e. many competing product states). Indeed, this is basically how nitrogenase chemists have long reasoned about the electronic structure of iron-sulfur clusters and FeMo-co (Lovell et al. 2001; Yamaguchi et al. 1990). Our work thus now provides extensive and rigorous numerical support for this picture.
Because of this simplicity, the full richness of classical quantum chemistry methods can now be brought to bear on FeMo-co electronic structure beyond the LLDUC model. Assuming the model already captures the qualitative complexity of the cluster’s electronic structure, we expect such investigations to provide quantitative corrections to the picture we have obtained. We took some initial steps to confirm this in our manuscript, considering larger orbital spaces, the effect of protein fluctuations, and the interpretation of certain spectroscopies. In the future, connecting these simulations to more spectroscopic measurements will be an exciting possibility. In addition, now that the electronic structure is on a conceptually sound footing, we have a foundation to support the central question of resolving the reaction mechanism. This opens up a whole new set of scientific challenges associated with observing reactions on extremely slow timescales.
Implications of the classical solution for quantum computing in chemistry
Because of the success of classical heuristic methods for this problem, one may naturally wonder what these results mean for the application of quantum computers in chemistry. Here I address some commonly asked questions.
Is the classical simulation of the LLDUC model a ‘last hurrah’ for classical methods?
I have seen the analogy drawn between the FeMo-co result and the classical tensor network simulations for random circuit sampling experiments. In that case, while the famous Google Sycamore experiment (Arute et al. 2019) could be replicated by classical tensor network simulations (Gray and Kourtis 2021), subsequent improvements in quantum processors, soon led to random circuit sampling experiments outpacing the capabilities of classical simulations.
However, the situation here is quite different. There is strong evidence that generating samples from a random quantum circuit (without noise) is actually exponentially hard to do using a classical algorithm, and indeed, the classical simulations used for the task were (mostly) brute force simulations with exponential cost in circuit size. In contrast, the theoretical support for exponential quantum advantage in the FeMo-co problem is much weaker, and as an empirical fact, most of the methods used in the FeMo-co simulation (namely the coupled cluster methods for a given excitation level) are polynomial cost algorithms. Since a similar simulation strategy has also been successfully applied across the series of 2, 4, and 8 metal iron-sulfur complexes (Sharma et al. 2014; Li et al. 2019; Zhai et al. 2023, 2026), we have no reason to expect a radically different situation if we consider larger analogous complexes in this series.
And in any case, chemistry does not provide an endless scaling of problem size; FeMo-co is the largest enzyme cofactor in terms of the number of transition metals. Materials simulations provide a setting to scale the problem size, but one still faces the question as to whether the relevant states observed are truly that complicated classically. For example, classical simulations of the ground-state orders of the 2D Hubbard model currently show no exponential increase in difficulty when going to larger system sizes (Chen et al. 2025; Liu et al. 2025).
Has the availability of a classical strategy for FeMo-co changed your enthusiasm for quantum computers in chemistry?
Again, my answer is no. There is an entire community of nitrogenase scientists: experimental spectroscopists, synthetic chemists, and of course computational chemists, who are working to map out the reaction mechanism, none of whose research is predicated on using a quantum computer. Personally, I have never thought that to understand nitrogenase we would first have to build a quantum computer, otherwise I would not work on the problem!
At the same time, any computational tool brings new capabilities that will be useful. Quantum algorithms come with theoretical guarantees; for example, so long as the initial state is well prepared, we know the error in the energy that we measure from a quantum algorithm, which is more reliable than the classical estimates of error we obtain from extrapolations. Similarly, initial state preparation for a quantum computer, even for classically tractable problems, is probably easier than solving the entire problem classically, since only a ‘rough’ guess is needed. And finally, a polynomial or even constant factor speedup is exciting, so long as the speedup is large enough!
Thus, I am in fact excited to see quantum computers applied to this problem, I am just not waiting for them to be built first.
How should one think about past work on quantum algorithms that has used FeMo-co as a target?
FeMo-co was amongst the earliest examples of a chemical problem for which a case for quantum advantage was made. For this reason, it is overrepresented in the literature of quantum computing for chemistry. Should fully fault-tolerant quantum computers be available, they will naturally be applied to a wider set of systems (Chan 2024; Babbush et al. 2025).
Also, one must recognize that the availability of a single concrete optimization target has led to undeniable advances in quantum algorithms for quantum chemistry. In most cases, prior work to improve quantum resources estimates for FeMo-co involve techniques that apply to other systems as well. Thus, there’s no need to throw away those papers!
What are some lessons and conclusions to draw?
The first is obviously that, just because something has not been solved, or appears hard to solve classically, does not mean it is the best problem to choose for a quantum computer. The classical solution strategy for FeMo-co essentially involved a complicated classical state preparation problem, which is a shared challenge with ground-state estimation algorithms in quantum computers, and thus not perhaps an optimal choice of problem.
My second main conclusion is that since classical solutions in complex problems are possible because they use some understanding of the problem, for quantum algorithms to have maximum impact, they should use the same knowledge. In fact most chemistry is not about truly mysterious quantum systems, but more about ordinary quantum matter where we know roughly what is going on, but where detailed simulations are still required. If quantum computing algorithms can target this ‘mundane’ regime, they will have maximum impact on chemistry as it is practiced today. In recent work, we have taken some steps in this direction by proposing quantum algorithms for electronic structure that work within the same heuristic framework as most current quantum chemistry methods (Chen and Chan 2025).
Finally, I wish to emphasize that, from the perspective of understanding nitrogenase, and maximising societal impact, the choice of computational algorithm and hardware to solve the problem is irrelevant. The fact that FeMo-co electronic structure is not so mysterious is an enormously positive thing, as it means that making progress on the larger problem of the mechanism using computation no longer seems so impossible. I have seen some of the brightest minds in the world helping to advance quantum algorithms for this problem. If any of this brainpower can be devoted to the chemical question itself, I believe we can be very optimistic about the future solution of the nitrogenase problem.





