
Do you know when an engineer built the first artificial automaton—the first human-made machine that operated by itself, without external control mechanisms that altered the machine’s behavior over time as the machine undertook its mission?
The ancient Greek thinker Archytas of Tarentum reportedly created it about 2,300 years ago. Steam propelled his mechanical pigeon through the air.

For centuries, automata cropped up here and there as curiosities and entertainment. The wealthy exhibited automata to amuse and awe their peers and underlings. For instance, the French engineer Jacques de Vauconson built a mechanical duck that appeared to eat and then expel grains. The device earned the nickname the Digesting Duck…and the nickname the Defecating Duck.

Vauconson also invented a mechanical loom that helped foster the Industrial Revolution. During the 18th and 19th centuries, automata began to enable factories, which changed the face of civilization. We’ve inherited the upshots of that change. Nowadays, cars drive themselves, Roombas clean floors, and drones deliver packages.1 Automata have graduated from toys to practical tools.2
Rather, classical automata have. What of their quantum counterparts?
Scientists have designed autonomous quantum machines, and experimentalists have begun realizing them. The roster of such machines includes autonomous quantum engines, refrigerators, and clocks. Much of this research falls under the purview of quantum thermodynamics, due to the roles played by energy in these machines’ functioning: above, I defined an automaton as a machine free of time-dependent control (exerted by a user). Equivalently, according to a thermodynamicist mentality, we can define an automaton as a machine on which no user performs any work as the machine operates. Thermodynamic work is well-ordered energy that can be harnessed directly to perform a useful task. Often, instead of receiving work, an automaton receives access to a hot environment and a cold environment. Heat flows from the hot to the cold, and the automaton transforms some of the heat into work.
Quantum automata appeal to me because quantum thermodynamics has few practical applications, as I complained in my previous blog post. Quantum thermodynamics has helped illuminate the nature of the universe, and I laud such foundational insights. Yet we can progress beyond laudation by trying to harness those insights in applications. Some quantum thermal machines—quantum batteries, engines, etc.—can outperform their classical counterparts, according to certain metrics. But controlling those machines, and keeping them cold enough that they behave quantum mechanically, costs substantial resources. The machines cost more than they’re worth. Quantum automata, requiring little control, offer hope for practicality.
To illustrate this hope, my group partnered with Simone Gasparinetti’s lab at Chalmer’s University in Sweden. The experimentalists created an autonomous quantum refrigerator from superconducting qubits. The quantum refrigerator can help reset, or “clear,” a quantum computer between calculations.

After we wrote the refrigerator paper, collaborators and I raised our heads and peered a little farther into the distance. What does building a useful autonomous quantum machine take, generally? Collaborators and I laid out guidelines in a “Key Issues Review” published in Reports in Progress on Physics last November.
We based our guidelines on DiVincenzo’s criteria for quantum computing. In 1996, David DiVincenzo published seven criteria that any platform, or setup, must meet to serve as a quantum computer. He cast five of the criteria as necessary and two criteria, related to information transmission, as optional. Similarly, our team provides ten criteria for building useful quantum automata. We regard eight of the criteria as necessary, at least typically. The final two, optional guidelines govern information transmission and machine transportation.

DiVincenzo illustrated his criteria with multiple possible quantum-computing platforms, such as ions. Similarly, we illustrate our criteria in two ways. First, we show how different quantum automata—engines, clocks, quantum circuits, etc.—can satisfy the criteria. Second, we illustrate how quantum automata can consist of different platforms: ultracold atoms, superconducting qubits, molecules, and so on.
Nature has suggested some of these platforms. For example, our eyes contain autonomous quantum energy transducers called photoisomers, or molecular switches. Suppose that such a molecule absorbs a photon. The molecule may use the photon’s energy to switch configuration. This switching sets off chemical and neurological reactions that result in the impression of sight. So the quantum switch transduces energy from light into mechanical, chemical, and electric energy.

My favorite of our criteria ranks among the necessary conditions: every useful quantum automata must produce output worth the input. How one quantifies a machine’s worth and cost depends on the machine and on the user. For example, an agent using a quantum engine may care about the engine’s efficiency, power, or efficiency at maximum power. Costs can include the energy required to cool the engine to the quantum regime, as well as the control required to initialize the engine. The agent also chooses which value they regard as an acceptable threshold for the output produced per unit input. I like this criterion because it applies a broom to dust that we quantum thermodynamicists often hide under a rug: quantum thermal machines’ costs. Let’s begin building quantum engines that perform more work than they require to operate.
One might object that scientists and engineers are already sweating over nonautonomous quantum machines. Companies, governments, and universities are pouring billions of dollars into quantum computing. Building a full-scale quantum computer by hook or by crook, regardless of classical control, is costing enough. Eliminating time-dependent control sounds even tougher. Why bother?
Fellow Quantum Frontiers blogger John Preskill pointed out one answer, when I described my new research program to him in 2022: control systems are classical—large and hot. Consider superconducting qubits—tiny quantum circuits—printed on a squarish chip about the size of your hand. A control wire terminates on each qubit. The rest of the wire runs off the edge of the chip, extending to classical hardware standing nearby. One can fit only so many wires on the chip, so one can fit only so many qubits. Also, the wires, being classical, are hotter than the qubits should be. The wires can help decohere the circuits, introducing errors into the quantum information they store. The more we can free the qubits from external control—the more autonomy we can grant them—the better.

Besides, quantum automata exemplify quantum steampunk, as my coauthor Pauli Erker observed. I kicked myself after he did, because I’d missed the connection. The irony was so thick, you could have cut it with the retractible steel knife attached to a swashbuckling villain’s robotic arm. Only two years before, I’d read The Watchmaker of Filigree Street, by Natasha Pulley. The novel features a Londoner expatriate from Meiji Japan, named Mori, who builds clockwork devices. The most endearing is a pet-like octopus, called Katsu, who scrambles around Mori’s workshop and hoards socks.

Does the world need a quantum version of Katsu? Not outside of quantum-steampunk fiction…yet. But a girl can dream. And quantum automata now have the opportunity to put quantum thermodynamics to work.

1And deliver pizzas. While visiting the University of Pittsburgh a few years ago, I was surprised to learn that the robots scurrying down the streets were serving hungry students.
2And minions of starving young scholars.













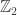 symmetry. The system has two phases, analogous to the liquid and ice phases of H
symmetry. The system has two phases, analogous to the liquid and ice phases of H O. Which phase the system occupies depends on the chemical potential—the average amount of energy needed to add a particle to the system (while the system’s entropy, its volume, and more remain constant).
O. Which phase the system occupies depends on the chemical potential—the average amount of energy needed to add a particle to the system (while the system’s entropy, its volume, and more remain constant).




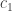 ,
, 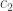 ,
, 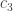 , and
, and 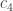 . How accurately can you predict the cell’s location? That is, what probability does the cell have of sitting at some particular site, conditioned on the
. How accurately can you predict the cell’s location? That is, what probability does the cell have of sitting at some particular site, conditioned on the  s? That probability is large only at one site, biophysicists have found empirically. So a cell can accurately infer its position from its molecules’ concentrations.
s? That probability is large only at one site, biophysicists have found empirically. So a cell can accurately infer its position from its molecules’ concentrations.


 denote the cold environment’s temperature; and
denote the cold environment’s temperature; and  , the hot environment’s. The efficiency can’t exceed
, the hot environment’s. The efficiency can’t exceed  . What a simple formula for such an extensive class of objects! Carnot’s theorem governs not only many car engines (Otto engines), but also the
. What a simple formula for such an extensive class of objects! Carnot’s theorem governs not only many car engines (Otto engines), but also the 










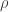 (about diverse observables’ expectation values). We have to measure many copies of
(about diverse observables’ expectation values). We have to measure many copies of 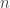 of copies. The community expected
of copies. The community expected 













